Documentary Proof: US Army Research Centers Generated COVID-19
This article contains hard proof that cannot be questioned or denied, which you may submit to any government agency or healthcare professional. What is not yet proven, but coming into focus, is that the US biological weapons program at Fort Detrick, Maryland, equipment and certainly key staff, migrated to secret labs at large state universities in order to “hide in plain sight.” Follow the careers of those who worked on the Wuhan-COVID project in 2017. Also, note that the exact same personnel and equipment is used for fake “prevention” research and testing as weaponization and actual production.
Submit this paper to any physician, or other qualified bio-sciences specialist. See what they say.
Introduction
Documents below will show that research to create COVID-19 began in the United States in 2006 and culminated in a successful bio-weapon in 2015, with work done at the University of North Carolina and at Harvard and at the Food and Drug Administration’s lab in Arkansas. Their work was titled: A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence.
COVID-19 was a US Army bio-weapons project to manufacture a pneumonia-causing disease that would be nearly impossible to vaccinate for in patients over 40 years old. The study was run by the University of North Carolina and funded by USAID/CIA. It chose a Chinese bat virus and chose to include a medical facility in Wuhan as well. Now we know why, a smokescreen of the blame for a program China had little or nothing to do with, something satanically evil and purely American.
In November 2015, a study was published outlining the capability of producing the virus we are dealing with now. Among the many involved was a lab in Wuhan, China. It was listed from the beginning as one of the dozens, mostly American, working on this project.
However, one key participant was left out, USAID. It is suspected, deeply so, that USAID is a front for American bio-warfare research such as that done in Tbilisi, Georgia, and elsewhere, much documented. This is the citation that adds USAID to the research funding group.
Change history
We will present Pravda’s biased article and, below that, the actual study proving the capability of producing COVID 19, proving it is not a naturally occurring virus once and for all.
As to who did what, this is not our job but we are proving, categorically, that when a Chinese lab is mentioned, it is a minor player in an American effort, as outlined exhaustively below. This makes discussing the Wuhan lab possibly complicit in bio-warfare.
Similarly, when Forbes Magazine and others stated they could prove COVID 19 was made naturally, and of course they had the same access we have, we suspect that they are part of a disinformation effort tied to USAID and bio-warfare.
Suspicion is not proof. The proof is proof and there is proof enough to drown in. The American medical professionals who pimped themselves out to the US Army and CIA helped bring US where it is now, a nation broken to pieces.
Pravda.ru:
“Such material appeared in 2015 on the website of the scientific journal Natura in 2015. Then the authors claimed that after the advent of the SARS virus (2002-2003) and the Middle East respiratory syndrome (MERS), scientists were aware of the risk of interspecific transmission that would lead to an epidemic among people.
Successful lab experiment
Among others, the research team studied bats, which are the largest incubators of coronaviruses. Nevertheless, bats could not transmit the coronavirus to humans because they could not interact with human cells with ACE2 receptors.
The material also stated that horseshoe bats carry a strain of SARS coronavirus that can be transmitted to humans. It has been named the SHC014-CoV virus.
To better study this virus, scientists copied the coronavirus and infected it with laboratory mice. The results showed that the virus is really able to bind to human cells with ACE2 receptors and multiply in the cells of the respiratory system.
In the research work, it is noted that laboratory materials, samples and equipment that were used in the research were obtained from the Army Medical Research Institute of Infectious Diseases. It is not yet possible to say for sure that the virus that was tested in laboratory mice is the same as the SARS-Cove-2 coronavirus.
NATO policy
However, interesting info can be found in earlier documents. For example:
• The 2019 Alliance’s activity report says that in 2019, the Alliance’s first place in research and development was occupied by the topic of radiochemical and biological protection (29%), shifting the seemingly most pressing problem of Europe – counterterrorism (it turned out to be 4th priority).
• A year earlier, in 2018, the situation was exactly the opposite: terrorism, as it should be, was in the first place (28%), and radiochemical and biological protection in the fourth (13%).
As the Brussels snitch writes in the telegram channel, “given the absence of visible reasons for such a sharp change in scientific interests, there are two options and both are unpleasant:
• or NATO now wags the fifth point, falsifying the data to show ‘we always prepared for viruses, we are modern’,
• or even in 2019 in the alliance, God forgive me, they knew where the trouble would come from.”
Yes, the first option is much more real, but, you see, the facts are surprising.”
Original 2015 Research Unedited and Complete
Published on 09 November 2015, A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence by Vineet D Menachery, Boyd L Yount Jr, Kari Debbink, Sudhakar Agnihothram, Lisa E Gralinski, Jessica A Plante, Rachel L Graham, Trevor Scobey, Xing-Yi Ge, Eric F Donaldson, Scott H Randell, Antonio Lanzavecchia, Wayne A Marasco, Zhengli-Li Shi, Ralph S Baric in Nature Medicine volume 21, pages1508–1513 (2015). A Corrigendum to this article was published on 06 April 2016.
Abstract
The emergence of severe acute respiratory syndrome coronavirus (SARS-CoV) and the Middle East respiratory syndrome (MERS)-CoV underscores the threat of cross-species transmission events leading to outbreaks in humans. Here we examine the disease potential of a SARS-like virus, SHC014-CoV, which is currently circulating in Chinese horseshoe bat populations. Using the SARS-CoV reverse genetics system, we generated and characterized a chimeric virus expressing the spike of bat coronavirus SHC014 in a mouse-adapted SARS-CoV backbone.
The results indicate that group 2b viruses encoding the SHC014 spike in a wild-type backbone can efficiently use multiple orthologs of the SARS receptor human angiotensin-converting enzyme II (ACE2), replicate efficiently in primary human airway cells and achieve in vitro titers equivalent to epidemic strains of SARS-CoV. Additionally, in vivo experiments demonstrate replication of the chimeric virus in mouse lung with notable pathogenesis.
Evaluation of available SARS-based immune-therapeutic and prophylactic modalities revealed poor efficacy; both monoclonal antibody and vaccine approach failed to neutralize and protect from infection with CoVs using the novel spike protein.
On the basis of these findings, we synthetically re-derived an infectious full-length SHC014 recombinant virus and demonstrate robust viral replication both in vitro and in vivo. Our work suggests a potential risk of SARS-CoV re-emergence from viruses currently circulating in bat populations.
Main
Note: All figures mentioned below can be found here and all supplementary figures and tables can be found here.
The emergence of SARS-CoV heralded a new era in the cross-species transmission of severe respiratory illness with globalization leading to rapid spread around the world and massive economic impact. Since then, several strains—including influenza A strains H5N1, H1N1 and H7N9, and MERS-CoV—have emerged from animal populations, causing considerable disease, mortality and economic hardship for the afflicted regions. Although public health measures were able to stop the SARS-CoV outbreak, recent metagenomics studies have identified sequences of closely related SARS-like viruses circulating in Chinese bat populations that may pose a future threat.
However, sequence data alone provides minimal insights to identify and prepare for future pre-pandemic viruses. Therefore, to examine the emergence potential (that is, the potential to infect humans) of circulating bat CoVs, we built a chimeric virus encoding a novel, zoonotic CoV spike protein—from the RsSHC014-CoV sequence that was isolated from Chinese horseshoe bats—in the context of the SARS-CoV mouse-adapted backbone. The hybrid virus allowed us to evaluate the ability of the novel spike protein to cause disease independently of other necessary adaptive mutations in its natural backbone.
Using this approach, we characterized CoV infection mediated by the SHC014 spike protein in primary human airway cells and in vivo and tested the efficacy of available immune therapeutics against SHC014-CoV. Together, the strategy translates metagenomics data to help predict and prepare for future emergent viruses.
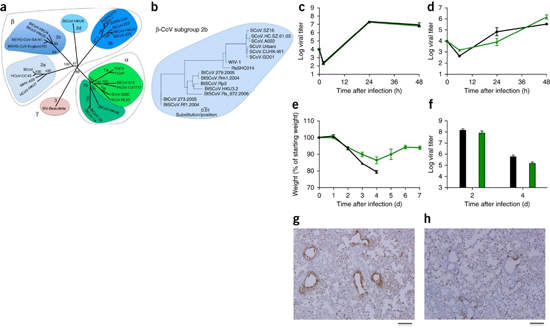
The sequences of SHC014 and the related RsWIV1-CoV show that these CoVs are the closest relatives to the epidemic SARS-CoV strains (Fig. 1a,b); however, there are important differences in the 14 residues that bind human ACE2, the receptor for SARS-CoV, including the five that are critical for host range: Y442, L472, N479, T487 and Y491.
In WIV1, three of these residues vary from the epidemic SARS-CoV Urbani strain, but they were not expected to alter binding to ACE2 (Supplementary Fig. 1a,b and Supplementary Table 1). This fact is confirmed by both pseudotyping experiments that measured the ability of lentiviruses encoding WIV1 spike proteins to enter cells expressing human ACE2 (Supplementary Fig. 1) and by in vitro replication assays of WIV1-CoV. In contrast, 7 of 14 ACE2-interaction residues in SHC014 are different from those in SARS-CoV, including all five residues critical for host range (Supplementary Fig. 1c and Supplementary Table 1).
These changes, coupled with the failure of pseudotyped lentiviruses expressing the SHC014 spike to enter cells (Supplementary Fig. 1d), suggested that the SHC014 spike is unable to bind human ACE2. However, similar changes in related SARS-CoV strains had been reported to allow ACE2 binding, suggesting that additional functional testing was required for verification.
Therefore, we synthesized the SHC014 spike in the context of the replication-competent, mouse-adapted SARS-CoV backbone (we hereafter refer to the chimeric CoV as SHC014-MA15) to maximize the opportunity for pathogenesis and vaccine studies in mice (Supplementary Fig. 2a). Despite predictions from both structure-based modeling and pseudotyping experiments, SHC014-MA15 was viable and replicated to high titers in Vero cells (Supplementary Fig. 2b). Similar to SARS, SHC014-MA15 also required a functional ACE2 molecule for entry and could use human, civet and bat ACE2 orthologs (Supplementary Fig. 2c,d).
To test the ability of the SHC014 spike to mediate infection of the human airway, we examined the sensitivity of the human epithelial airway cell line Calu-3 2B4 to infection and found robust SHC014-MA15 replication, comparable to that of SARS-CoV Urbani (Fig. 1c). To extend these findings, primary human airway epithelial (HAE) cultures were infected and showed robust replication of both viruses (Fig. 1d). Together, the data confirm the ability of viruses with the SHC014 spike to infect human airway cells and underscore the potential threat of cross-species transmission of SHC014-CoV.
Full size image
Figure 1: SARS-like viruses replicate in human airway cells and produce in vivo pathogenesis.
(a) The full-length genome sequences of representative CoVs were aligned and phylogenetically mapped as described in the Online Methods. The scale bar represents nucleotide substitutions, with only bootstrap support above 70% being labeled. The tree shows CoVs divided into three distinct phylogenetic groups, defined as α-CoVs, β-CoVs and γ-CoVs. Classical subgroup clusters are marked as 2a, 2b, 2c and 2d for the β-CoVs and as 1a and 1b for the α-CoVs. (b) Amino acid sequences of the S1 domains of the spikes of representative β-CoVs of the 2b group, including SARS-CoV, were aligned and phylogenetically mapped. The scale bar represents the amino acid substitutions. (c,d) Viral replication of SARS-CoV Urbani (black) and SHC014-MA15 (green) after infection of Calu-3 2B4 cells (c) or well-differentiated, primary air-liquid interface HAE cell cultures (d) at a multiplicity of infection (MOI) of 0.01 for both cell types. Samples were collected at individual time points with biological replicates (n = 3) for both Calu-3 and HAE experiments. (e,f) Weight loss (n = 9 for SARS-CoV MA15; n = 16 for SHC014-MA15) (e) and viral replication in the lungs (n = 3 for SARS-CoV MA15; n = 4 for SHC014-MA15) (f) of 10-week-old BALB/c mice infected with 1 × 104 p.f.u. of mouse-adapted SARS-CoV MA15 (black) or SHC014-MA15 (green) via the intranasal (i.n.) route. (g,h) Representative images of lung sections stained for SARS-CoV N antigen from mice infected with SARS-CoV MA15 (n = 3 mice) (g) or SHC014-MA15 (n = 4 mice) (h) are shown. For each graph, the center value represents the group mean, and the error bars define the s.e.m. Scale bars, 1 mm.
To evaluate the role of the SHC014 spike in mediating infection in vivo, we infected 10-week-old BALB/c mice with 104 plaque-forming units (p.f.u.) of either SARS-MA15 or SHC014-MA15 (Fig. 1e–h). Animals infected with SARS-MA15 experienced rapid weight loss and lethality by 4 d post-infection (d.p.i.); in contrast, SHC014-MA15 infection produced substantial weight loss (10%) but no lethality in mice (Fig. 1e). Examination of viral replication revealed nearly equivalent viral titers from the lungs of mice infected with SARS-MA15 or SHC014-MA15 (Fig. 1f). Whereas lungs from the SARS-MA15–infected mice showed robust staining in both the terminal bronchioles and the lung parenchyma 2 d.p.i. (Fig. 1g), those of SHC014-MA15–infected mice showed reduced airway antigen staining (Fig. 1h); in contrast, no deficit in antigen staining was observed in the parenchyma or in the overall histology scoring, suggesting differential infection of lung tissue for SHC014-MA15 (Supplementary Table 2).
We next analyzed infection in more susceptible, aged (12-month-old) animals. SARS-MA15–infected animals rapidly lost weight and succumbed to infection (Supplementary Fig. 3a,b). SHC014-MA15 infection-induced robust and sustained weight loss, but had minimal lethality. Trends in the histology and antigen staining patterns that we observed in young mice were conserved in the older animals (Supplementary Table 3). We excluded the possibility that SHC014-MA15 was mediating infection through an alternative receptor on the basis of experiments using Ace2−/− mice, which did not show weight loss or antigen staining after SHC014-MA15 infection (Supplementary Fig. 4a,b and Supplementary Table 2). Together, the data indicate that viruses with the SHC014 spike are capable of inducing weight loss in mice in the context of a virulent CoV backbone.
Given the preclinical efficacy of Ebola monoclonal antibody therapies, such as ZMApp10, we next sought to determine the efficacy of SARS-CoV monoclonal antibodies against infection with SHC014-MA15. Four broadly neutralizing human monoclonal antibodies targeting SARS-CoV spike protein had been previously reported and are probable reagents for immunotherapy. We examined the effect of these antibodies on viral replication (expressed as percentage inhibition of viral replication) and found that whereas wild-type SARS-CoV Urbani was strongly neutralized by all four antibodies at relatively low antibody concentrations (Fig. 2a–d), neutralization varied for SHC014-MA15. Fm6, an antibody generated by phage display and escape mutants, achieved only background levels of inhibition of SHC014-MA15 replication (Fig. 2a). Similarly, antibodies 230.15 and 227.14, which were derived from memory B cells of SARS-CoV–infected patients, also failed to block SHC014-MA15 replication (Fig. 2b,c). For all three antibodies, differences between the SARS and SHC014 spike amino acid sequences corresponded to direct or adjacent residue changes found in SARS-CoV escape mutants (fm6 N479R; 230.15 L443V; 227.14 K390Q/E), which probably explains the absence of the antibodies’ neutralizing activity against SHC014. Finally, monoclonal antibody 109.8 was able to achieve 50% neutralization of SHC014-MA15, but only at high concentrations (10 μg/ml) (Fig. 2d). Together, the results demonstrate that broadly neutralizing antibodies against SARS-CoV may only have marginal efficacy against emergent SARS-like CoV strains such as SHC014.
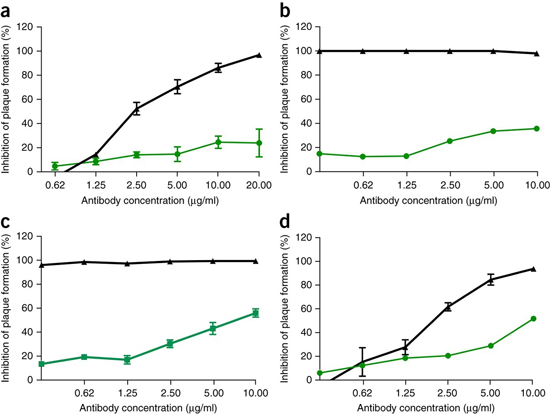
Full size image
Figure 2: SARS-CoV monoclonal antibodies have marginal efficacy against SARS-like CoVs.
(a–d) Neutralization assays evaluating efficacy (measured as a reduction in the number of plaques) of a panel of monoclonal antibodies, which were all originally generated against epidemic SARS-CoV, against infection of Vero cells with SARS-CoV Urbani (black) or SHC014-MA15 (green). The antibodies tested were fm6 (n = 3 for Urbani; n = 5 for SHC014-MA15)11,12 (a), 230.15 (n = 3 for Urbani; n = 2 for SHC014-MA15) (b), 227.15 (n = 3 for Urbani; n = 5 for SHC014-MA15) (c) and 109.8 (n = 3 for Urbani; n = 2 for SHC014-MA15)13 (d). Each data point represents the group mean and error bars define the s.e.m. Note that the error bars in SARS-CoV Urbani–infected Vero cells in b,c are overlapped by the symbols and are not visible.
To evaluate the efficacy of existing vaccines against infection with SHC014-MA15, we vaccinated aged mice with double-inactivated whole SARS-CoV (DIV). Previous work showed that DIV could neutralize and protect young mice from challenge with a homologous virus; however, the vaccine failed to protect aged animals in which augmented immune pathology was also observed, indicating the possibility of the animals being harmed because of the vaccination. Here we found that DIV did not provide protection from challenge with SHC014-MA15 with regards to weight loss or viral titer (Supplementary Fig. 5a,b). Consistent with a previous report with other heterologous groups 2b CoVs15, serum from DIV-vaccinated, aged mice also failed to neutralize SHC014-MA15 (Supplementary Fig. 5c). Notably, DIV vaccination resulted in robust immune pathology (Supplementary Table 4) and eosinophilia (Supplementary Fig. 5d–f). Together, these results confirm that the DIV vaccine would not be protective against infection with SHC014 and could possibly augment disease in the aged vaccinated group.
In contrast to the vaccination of mice with DIV, the use of SHC014-MA15 as a live, attenuated vaccine showed potential cross-protection against challenge with SARS-CoV, but the results have important caveats. We infected young mice with 104 p.f.u. of SHC014-MA15 and observed them for 28 d. We then challenged the mice with SARS-MA15 at day 29 (Supplementary Fig. 6a). The prior infection of the mice with the high dose of SHC014-MA15 conferred protection against challenge with a lethal dose of SARS-MA15, although there was only a minimal SARS-CoV neutralization response from the antisera elicited 28 d after SHC014-MA15 infection (Supplementary Fig. 6b, 1:200). In the absence of a secondary antigen boost, 28 d.p.i. represents the expected peak of antibody titers and implies that there will be diminished protection against SARS-CoV over time.
Similar results showing protection against challenge with a lethal dose of SARS-CoV were observed in aged BALB/c mice with respect to weight loss and viral replication (Supplementary Fig. 6c,d). However, the SHC014-MA15 infection dose of 104 p.f.u. induced >10% weight loss and lethality in some aged animals (Fig. 1 and Supplementary Fig. 3). We found that vaccination with a lower dose of SHC014-MA15 (100 p.f.u.), did not induce weight loss, but it also failed to protect aged animals from a SARS-MA15 lethal dose challenge (Supplementary Fig. 6e,f). Together, the data suggest that SHC014-MA15 challenge may confer cross-protection against SARS-CoV through conserved epitopes, but the required dose induces pathogenesis and precludes use as an attenuated vaccine.
Having established that the SHC014 spike has the ability to mediate infection of human cells and cause disease in mice, we next synthesized a full-length SHC014-CoV infectious clone based on the approach used for SARS-CoV (Fig. 3a). Replication in Vero cells revealed no deficit for SHC014-CoV relative to that for SARS-CoV (Fig. 3b); however, SHC014-CoV was significantly (P<0.01) attenuated in primary HAE cultures at both 24 and 48 h after infection (Fig. 3c). In vivo infection of mice demonstrated no significant weight loss but showed reduced viral replication in lungs of full-length SHC014-CoV infection, as compared to SARS-CoV Urbani (Fig. 3d,e). Together, the results establish the viability of full-length SHC014-CoV, but suggest that further adaptation is required for its replication to be equivalent to that of epidemic SARS-CoV in human respiratory cells and in mice.
Full size image
Figure 3: Full-length SHC014-CoV replicates in human airways but lacks the virulence of epidemic SARS-CoV.
(a) Schematic of the SHC014-CoV molecular clone, which was synthesized as six contiguous cDNAs (designated SHC014A, SHC014B, SHC014C, SHC014D, SHC014E and SHC014F) flanked by unique BglI sites that allowed for directed assembly of the full-length cDNA expressing open reading frames (for 1a, 1b, spike, 3, envelope, matrix, 6–8 and nucleocapsid). Underlined nucleotides represent the overhang sequences formed after restriction enzyme cleavage. (b,c) Viral replication of SARS-CoV Urbani (black) or SHC014-CoV (green) after infection of Vero cells (b) or well-differentiated, primary air-liquid interface HAE cell cultures (c) at an MOI of 0.01. Samples were collected at individual time points with biological replicates (n = 3) for each group. Data represent one experiment for both Vero and HAE cells. (d,e) Weight loss (n = 3 for SARS-CoV MA15, n = 7 for SHC014-CoV; n = 6 for SARS-Urbani) (d) and viral replication in the lungs (n = 3 for SARS-Urbani and SHC014-CoV) (e) of 10-week-old BALB/c mice infected with 1 × 105 p.f.u. of SARS-CoV MA15 (gray), SHC014-CoV (green) or SARS-CoV Urbani (black) via the i.n. route. Each data point represents the group mean, and error bars define the s.e.m. **P < 0.01 and ***P < 0.001 using two-tailed Student’s t-test of individual time points.
During the SARS-CoV epidemic, links were quickly established between palm civets and the CoV strains that were detected in humans. Building on this finding, the common emergence paradigm argues that epidemic SARS-CoV originated as a bat virus, jumped to civets, and incorporated changes within the receptor-binding domain (RBD) to improve binding to civet Ace2. Subsequent exposure to people in live-animal markets permitted human infection with the civet strain, which, in turn, adapted to become the epidemic strain (Fig. 4a). However, phylogenetic analysis suggests that early human SARS strains appear more closely related to bat strains than to civet strains. Therefore, a second paradigm argues that direct bat-human transmission initiated SARS-CoV emergence and that palm civets served as a secondary host and reservoir for continued infection (Fig. 4b). For both paradigms, spike adaptation in a secondary host is seen as a necessity, with most mutations expected to occur within the RBD, thereby facilitating improved infection. Both theories imply that pools of bat CoVs are limited and that host-range mutations are both random and rare, reducing the likelihood of future emergence events in humans.
Full size image
Figure 4: Emergence paradigms for coronaviruses.
Coronavirus strains are maintained in quasi-species pools circulating in bat populations. (a,b) Traditional SARS-CoV emergence theories posit that host-range mutants (red circle) represent random and rare occurrences that permit infection of alternative hosts. The secondary-host paradigm (a) argues that a nonhuman host is infected by a bat progenitor virus and, through adaptation, facilitates transmission to humans; subsequent replication in humans leads to the epidemic viral strain. The direct paradigm (b) suggests that transmission occurs between bats and humans without the requirement of an intermediate host; selection then occurs in the human population with closely related viruses replicating in a secondary host, permitting continued viral persistence and adaptation in both. (c) The data from chimeric SARS-like viruses argue that the quasi-species pools maintain multiple viruses capable of infecting human cells without the need for mutations (red circles). Although adaptations in secondary or human hosts may be required for epidemic emergence, if SHC014 spike–containing viruses recombined with virulent CoV backbones (circles with green outlines), then epidemic disease may be the result in humans. Existing data support elements of all three paradigms.
Although our study does not invalidate the other emergence routes, it does argue for a third paradigm in which circulating bat CoV pools maintain ‘poised’ spike proteins that are capable of infecting humans without mutation or adaptation (Fig. 4c). This hypothesis is illustrated by the ability of a chimeric virus containing the SHC014 spike in a SARS-CoV backbone to cause robust infection in both human airway cultures and in mice without RBD adaptation.
Coupled with the observation of previously identified pathogenic CoV backbones, our results suggest that the starting materials required for SARS-like emergent strains are currently circulating in animal reservoirs. Notably, although full-length SHC014-CoV probably requires additional backbone adaption to mediate human disease, the documented high-frequency recombination events in CoV families underscores the possibility of future emergence and the need for further preparation.
To date, genomics screens of animal populations have primarily been used to identify novel viruses in outbreak settings. The approach here extends these data sets to examine questions of viral emergence and therapeutic efficacy. We consider viruses with the SHC014 spike a potential threat owing to their ability to replicate in primary human airway cultures, the best available model for human disease. In addition, the observed pathogenesis in mice indicates a capacity for SHC014-containing viruses to cause disease in mammalian models, without RBD adaptation.
Notably, differential tropism in the lung as compared to that with SARS-MA15 and attenuation of full-length SHC014-CoV in HAE cultures relative to SARS-CoV Urbani suggest that factors beyond ACE2 binding—including spike processivity, receptor bio-availability or antagonism of the host immune responses—may contribute to emergence. However, further testing in nonhuman primates is required to translate these findings into the pathogenic potential in humans.
Importantly, the failure of available therapeutics defines a critical need for further study and for the development of treatments. With this knowledge, surveillance programs, diagnostic reagents, and effective treatments can be produced that are protective against the emergence of group 2b–specific CoVs, such as SHC014, and these can be applied to other CoV branches that maintain similarly heterogeneous pools.
In addition to offering preparation against future emerging viruses, this approach must be considered in the context of the US government-mandated pause on gain-of-function (GOF) studies.
On the basis of previous models of emergence (Fig. 4a,b), the creation of chimeric viruses such as SHC014-MA15 was not expected to increase pathogenicity. Although SHC014-MA15 is attenuated relative to its parental mouse-adapted SARS-CoV, similar studies examining the pathogenicity of CoVs with the wild-type Urbani spike within the MA15 backbone showed no weight loss in mice and reduced viral replication. Thus, relative to the Urbani spike–MA15 CoV, SHC014-MA15 shows a gain in pathogenesis (Fig. 1).
On the basis of these findings, scientific review panels may deem similar studies building chimeric viruses based on circulating strains too risky to pursue, as increased pathogenicity in mammalian models cannot be excluded.
Coupled with restrictions on mouse-adapted strains and the development of monoclonal antibodies using escape mutants, research into CoV emergence and therapeutic efficacy may be severely limited moving forward. Together, these data and restrictions represent a crossroads of GOF research concerns; the potential to prepare for and mitigate future outbreaks must be weighed against the risk of creating more dangerous pathogens. In developing policies moving forward, it is important to consider the value of the data generated by these studies and whether these types of chimeric virus studies warrant further investigation versus the inherent risks involved.
Overall, our approach has used metagenomics data to identify a potential threat posed by the circulating bat SARS-like CoV SHC014. Because of the ability of chimeric SHC014 viruses to replicate in human airway cultures, cause pathogenesis in vivo and escape current therapeutics, there is a need for both surveillance and improved therapeutics against circulating SARS-like viruses. Our approach also unlocks the use of metagenomics data to predict viral emergence and to apply this knowledge in preparing to treat future emerging virus infections.
Methods

Viruses, cells, in vitro infection and plaque assays
Wild-type SARS-CoV (Urbani), mouse-adapted SARS-CoV (MA15) and chimeric SARS-like CoVs were cultured on Vero E6 cells (obtained from United States Army Medical Research Institute of Infectious Diseases), grown in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, CA) and 5% fetal clone serum (FCS) (Hyclone, South Logan, UT) along with antibiotic/antimycotic (Gibco, Carlsbad, CA). DBT cells (Baric laboratory, source unknown) expressing ACE2 orthologs have been previously described for both human and civet; bat Ace2 sequence was based on that from Rhinolophus leschenaulti, and DBT cells expressing bat Ace2 were established as described previously.
Pseudotyping experiments were similar to those using an HIV-based pseudovirus, prepared as previously described10, and examined on HeLa cells (Wuhan Institute of Virology) that expressed ACE2 orthologs. HeLa cells were grown in minimal essential medium (MEM) (Gibco, CA) supplemented with 10% FCS (Gibco, CA) as previously described.
Growth curves in Vero E6, DBT, Calu-3 2B4, and primary human airway epithelial cells were performed as previously described. None of the working cell line stocks were authenticated or tested for mycoplasma recently, although the original seed stocks used to create the working stocks are free from contamination. Human lungs for HAE cultures were procured under the University of North Carolina at Chapel Hill Institutional Review Board–approved protocols. HAE cultures represent highly differentiated human airway epithelium containing ciliated and non-ciliated epithelial cells as well as goblet cells. The cultures are also grown on an air-liquid interface for several weeks before use, as previously described.
Briefly, cells were washed with PBS and inoculated with the virus or mock-diluted in PBS for 40 min at 37°C. After inoculation, cells were washed three times and a fresh medium was added to signify time ‘0’. Three or more biological replicates were harvested at each described time point. No blinding was used in any sample collections nor was samples randomized. All virus cultivation was performed in a biosafety level (BSL) 3 laboratory with redundant fans in the biosafety cabinets, as described previously by our group. All personnel wore powered air-purifying respirators (Breathe Easy, 3M) with Tyvek suits, aprons, and booties and were double-gloved.
Sequence clustering and structural modeling
The full-length genomic sequences and the amino acid sequences of the S1 domains of the spike of representative CoVs were downloaded from Genbank or Pathosystems Resource Integration Center (PATRIC), aligned with ClustalX and phylogenetically compared by using maximum likelihood estimation using 100 bootstraps or by using the PhyML package, respectively. The tree was generated using maximum likelihood with the PhyML package. The scale bar represents nucleotide substitutions. Only nodes with bootstrap support above 70% are labeled.
The tree shows that CoVs are divided into three distinct phylogenetic groups defined as α-CoVs, β-CoVs, and γ-CoVs. Classical subgroup clusters are marked as 2a, 2b, 2c, and 2d for β-CoVs, and 1a and 1b for the α-CoVs. Structural models were generated using Modeller (Max Planck Institute Bioinformatics Toolkit) to generate homology models for SHC014 and Rs3367 of the SARS RBD in complex with ACE2 based on crystal structure 2AJF (Protein Data Bank). Homology models were visualized and manipulated in MacPyMol (version 1.3).
Construction of SARS-like chimeric viruses
Both wild-type and chimeric viruses were derived from either SARS-CoV Urbani or the corresponding mouse-adapted (SARS-CoV MA15) infectious clone (ic) as previously described. Plasmids containing spike sequences for SHC014 were extracted by restriction digest and ligated into the E and F plasmid of the MA15 infectious clone. The clone was designed and purchased from Bio Basic as six contiguous cDNAs using published sequences flanked by unique class II restriction endonuclease sites (BglI). Thereafter, plasmids containing wild-type, chimeric SARS-CoV, and SHC014-CoV genome fragments were amplified, excised, ligated, and purified.
In vitro transcription reactions were then performed to synthesize full-length genomic RNA, which was transfected into Vero E6 cells as previously described. The medium from transfected cells was harvested and served as seed stocks for subsequent experiments. Chimeric and full-length viruses were confirmed by sequence analysis before use in these studies. Synthetic construction of chimeric mutant and full-length SHC014-CoV was approved by the University of North Carolina Institutional Biosafety Committee and the Dual Use Research of Concern committee.
Ethics statement
This study was carried out in accordance with the recommendations for the care and use of animals by the Office of Laboratory Animal Welfare (OLAW), NIH. The Institutional Animal Care and Use Committee (IACUC) of The University of North Carolina at Chapel Hill (UNC, Permit Number A-3410-01) approved the animal study protocol (IACUC #13-033) used in these studies.

Mice and in vivo infection
Female, 10-week-old, and 12-month-old BALB/cAnNHsD mice were ordered from Harlan Laboratories. Mouse infections were done as previously described. Briefly, animals were brought into a BSL3 laboratory and allowed to acclimate for 1 week before infection. For infection and live-attenuated virus vaccination, mice were anesthetized with a mixture of ketamine and xylazine and infected intranasally, when challenged, with 50 μl of phosphate-buffered saline (PBS) or diluted virus with three or four mice per time point, per infection group per dose as described in the figure legends.
For individual mice, notations for infection including failure to inhale the entire dose, bubbling of inoculum from the nose, or infection through the mouth may have led to exclusion of mouse data at the discretion of the researcher; post-infection, no other pre-established exclusion or inclusion criteria are defined. No blinding was used in any animal experiments, and animals were not randomized. For vaccination, young and aged mice were vaccinated by footpad injection with a 20-μl volume of either 0.2 μg of double-inactivated SARS-CoV vaccine with alum or mock PBS; mice were then boosted with the same regimen 22 d later and challenged 21 d thereafter. For all groups, as per protocol, animals were monitored daily for clinical signs of disease (hunching, ruffled fur, and reduced activity) for the duration of the experiment. Weight loss was monitored daily for the first 7 d, after which weight monitoring continued until the animals recovered to their initial starting weight or displayed weight gain continuously for 3 d.
All mice that lost greater than 20% of their starting body weight were ground-fed and further monitored multiple times per day as long as they were under the 20% cutoff. Mice that lost greater than 30% of their starting body weight were immediately sacrificed as per protocol. Any mouse deemed to be moribund or unlikely to recover was also humanely sacrificed at the discretion of the researcher. Euthanasia was performed using an isoflurane overdose and death was confirmed by cervical dislocation. All mouse studies were performed at the University of North Carolina (Animal Welfare Assurance #A3410-01) using protocols approved by the UNC Institutional Animal Care and Use Committee (IACUC).
Histological analysis
The left lung was removed and submerged in 10% buffered formalin (Fisher) without inflation for 1 week. Tissues were embedded in paraffin and 5-μm sections were prepared by the UNC Lineberger Comprehensive Cancer Center histopathology core facility. To determine the extent of antigen staining, sections were stained for viral antigen using a commercially available polyclonal SARS-CoV anti-nucleocapsid antibody (Imgenex) and scored in a blinded manner by for staining of the airway and parenchyma as previously described20. Images were captured using an Olympus BX41 microscope with an Olympus DP71 camera.
Virus neutralization assays
Plaque reduction neutralization titer assays were performed with previously characterized antibodies against SARS-CoV, as previously described. Briefly, neutralizing antibodies or serum was serially diluted twofold and incubated with 100 p.f.u. of the different infectious clone, SARS-CoV strains for 1 h at 37 °C. The virus and antibodies were then added to a 6-well plate with 5 × 105 Vero E6 cells/well with multiple replicates (n ≥ 2). After a 1-h incubation at 37°C, cells were overlaid with 3 ml of 0.8% agarose in a medium. Plates were incubated for 2 d at 37 °C, stained with neutral red for 3 h, and plaques were counted. The percentage of plaque reduction was calculated as (1 − (no. of plaques with an antibody/no. of plaques without antibody)) × 100.
Statistical analysis
All experiments were conducted contrasting two experimental groups (either two viruses, or vaccinated and unvaccinated cohorts). Therefore, significant differences in viral titer and histology scoring were determined by a two-tailed Student’s t-test at individual time points. Data were normally distributed in each group being compared and had a similar variance.
Biosafety and biosecurity
Reported studies were initiated after the University of North Carolina Institutional Biosafety Committee approved the experimental protocol (Project title: Generating infectious clones of bat SARS-like CoVs; Lab Safety Plan ID: 20145741; Schedule G ID: 12279).
These studies were initiated before the US Government Deliberative Process Research Funding Pause on Selected Gain-of-Function Research Involving Influenza, MERS, and SARS Viruses. This paper has been reviewed by the funding agency, the NIH. Continuation of these studies was requested, and this has been approved by the NIH.
SARS-CoV is a select agent. All work for these studies was performed with approved standard operating procedures (SOPs) and safety conditions for SARS-CoV, MERs-CoV, and other related CoVs. Our institutional CoV BSL3 facilities have been designed to conform to the safety requirements that are recommended in the Biosafety in Microbiological and Biomedical Laboratories (BMBL), the US Department of Health and Human Services, the Public Health Service, the Centers for Disease Control (CDC) and the NIH. Laboratory safety plans were submitted to, and the facility has been approved for use by, the UNC Department of Environmental Health and Safety (EHS) and the CDC. Electronic card access is required for entry into the facility.
All workers have been trained by EHS to safely use powered air-purifying respirators (PAPRs), and appropriate work habits in a BSL3 facility and active medical surveillance plans are in place. Our CoV BSL3 facilities contain redundant fans, emergency power to fans and biological safety cabinets and freezers, and our facilities can accommodate SealSafe mouse racks. Materials classified as BSL3 agents consist of SARS-CoV, bat CoV precursor strains, MERS-CoV and mutants derived from these pathogens. Within the BSL3 facilities, experimentation with an infectious virus is performed in a certified Class II Biosafety Cabinet (BSC).
All members of the staff wear scrubs, Tyvek suits and aprons, PAPRs and shoe covers, and their hands are double-gloved. BSL3 users are subject to a medical surveillance plan monitored by the University Employee Occupational Health Clinic (UEOHC), which includes a yearly physical, annual influenza vaccination and mandatory reporting of any symptoms associated with CoV infection during periods when working in the BSL3. All BSL3 users are trained in exposure management and reporting protocols, are prepared to self-quarantine and have been trained for safe delivery to a local infectious disease management department in an emergency situation. All potential exposure events are reported and investigated by EHS and UEOHC, with reports filed to both the CDC and the NIH.
yogaesoteric
July 17, 2020